ИИ ускорил моделирование молекул в 10 тыс. раз
Шведские ученые создали модель TITO, которая предсказывает движение молекул намного быстрее обычных расчетов. В будущем такой подход может ускорить ранний поиск новых лекарств.
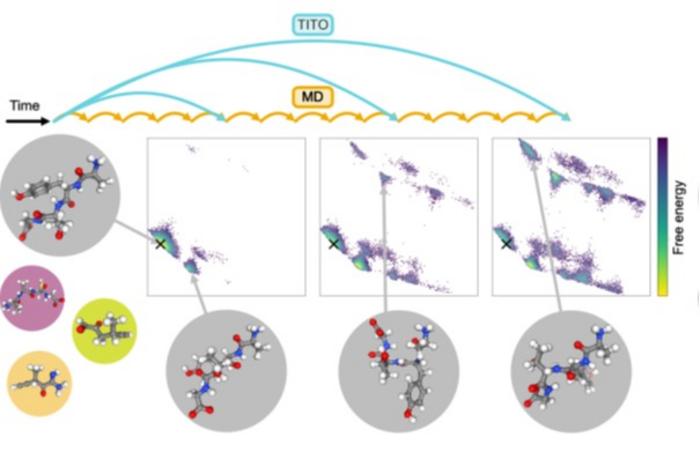
Источник: Juan Viguera Diez and Simon Olsson
МОСКВА, 12 июня. /Новости науки/. Искусственный интеллект помог ускорить моделирование движения молекул более чем в 10 тыс. раз по сравнению с обычными численными расчетами. . Работа опубликована в журнале Science Advances, сообщила пресс-служба Технического университета Чалмерса.
Обычное моделирование молекулярной динамики требует огромных вычислений. Компьютер должен шаг за шагом считать силы между атомами и каждый раз сдвигать их на очень малое расстояние. Один такой шаг длится около одной фемтосекунды, то есть 10⁻¹⁵ секунды. Но процессы, важные для разработки лекарств, идут намного дольше. Поэтому для одного расчета могут потребоваться миллиарды шагов.
Новая модель TITO, или Transferable Implicit Transfer Operators, учится не считать каждый шаг отдельно. Она анализирует примеры движения атомов и выводит общие правила, по которым меняется молекула. После этого модель может как бы «перематывать» молекулярный фильм вперед и сразу предсказывать, какие формы примет молекула и через какие переходы она пройдет.
«Наша модель отличается тем, что учится базовой динамике на более длинных временных масштабах. Она дает представление не только о формах молекул, но и о том, как быстро и какими путями происходят переходы», — сказал руководитель исследования, доцент Симон Олссон.
Ученые проверили модель более чем на 12,5 тыс. органических молекул, в том числе соединениях углерода, азота, водорода и кислорода. Также они изучили более тысячи коротких пептидов — небольших цепочек аминокислот, из которых состоят белки. Модель смогла предсказывать поведение молекул, которых не видела во время обучения. Это важно, потому что она не просто запоминает отдельные примеры, а находит общие закономерности движения атомов.
Исследователи сравнили выводы TITO с результатами обычных численных алгоритмов. По словам авторов, результаты согласуются друг с другом. При этом ИИ предсказывал изменения, которые происходят на временных отрезках примерно в тысячу раз длиннее, чем те, что он видел при обучении.
Главная возможная область применения — ранние этапы разработки лекарств. Сейчас создание нового препарата часто занимает более десяти лет. Большая часть времени и расходов приходится на первые проверки, когда ученые отсеивают тысячи молекул и ищут самые перспективные. Более быстрые расчеты могут помочь раньше понять, как молекула ведет себя в растворе, насколько она устойчива и может ли взаимодействовать с биологическими структурами.
Авторы подчеркивают, что технология пока не готова заменить лабораторные испытания. Сейчас метод проверили на малых молекулярных системах, в упрощенных моделях растворителя и при заданной температуре. Следующий этап — адаптация модели к более сложным и реалистичным системам.
Обычное моделирование молекулярной динамики требует огромных вычислений. Компьютер должен шаг за шагом считать силы между атомами и каждый раз сдвигать их на очень малое расстояние. Один такой шаг длится около одной фемтосекунды, то есть 10⁻¹⁵ секунды. Но процессы, важные для разработки лекарств, идут намного дольше. Поэтому для одного расчета могут потребоваться миллиарды шагов.
Новая модель TITO, или Transferable Implicit Transfer Operators, учится не считать каждый шаг отдельно. Она анализирует примеры движения атомов и выводит общие правила, по которым меняется молекула. После этого модель может как бы «перематывать» молекулярный фильм вперед и сразу предсказывать, какие формы примет молекула и через какие переходы она пройдет.
«Наша модель отличается тем, что учится базовой динамике на более длинных временных масштабах. Она дает представление не только о формах молекул, но и о том, как быстро и какими путями происходят переходы», — сказал руководитель исследования, доцент Симон Олссон.
Ученые проверили модель более чем на 12,5 тыс. органических молекул, в том числе соединениях углерода, азота, водорода и кислорода. Также они изучили более тысячи коротких пептидов — небольших цепочек аминокислот, из которых состоят белки. Модель смогла предсказывать поведение молекул, которых не видела во время обучения. Это важно, потому что она не просто запоминает отдельные примеры, а находит общие закономерности движения атомов.
Исследователи сравнили выводы TITO с результатами обычных численных алгоритмов. По словам авторов, результаты согласуются друг с другом. При этом ИИ предсказывал изменения, которые происходят на временных отрезках примерно в тысячу раз длиннее, чем те, что он видел при обучении.
Главная возможная область применения — ранние этапы разработки лекарств. Сейчас создание нового препарата часто занимает более десяти лет. Большая часть времени и расходов приходится на первые проверки, когда ученые отсеивают тысячи молекул и ищут самые перспективные. Более быстрые расчеты могут помочь раньше понять, как молекула ведет себя в растворе, насколько она устойчива и может ли взаимодействовать с биологическими структурами.
Авторы подчеркивают, что технология пока не готова заменить лабораторные испытания. Сейчас метод проверили на малых молекулярных системах, в упрощенных моделях растворителя и при заданной температуре. Следующий этап — адаптация модели к более сложным и реалистичным системам.



















